AMBER
Amber
Versions Installed
Kay: 16 / 18
Description
Amber (Assisted Model Building with Energy Refinement), is a general purpose molecular mechanics/dynamics suite which uses analytic potential energy functions, derived from experimental and ab initio data, to refine macromolecular conformations.
License
ICHEC has acquired a site license for the Amber 16 (AmberTools 17) and Amber 18 (AmberTools 18) packages.
Thin Component
A job submission example is as follows.
#!/bin/sh # All the informations about queues can be obtained by 'sinfo' # PARTITION AVAIL TIMELIMIT # DevQ up 1:00:00 # ProdQ* up 3-00:00:00 # LongQ up 6-00:00:00 # ShmemQ up 3-00:00:00 # PhiQ up 1-00:00:00 # GpuQ up 2-00:00:00 # Slurm flags #SBATCH -p ProdQ #SBATCH -N 2 #SBATCH --job-name=jobname #SBATCH -t 60:00:00 # Charge job to myaccount #SBATCH -A your_project # Write stdout+stderr to file #SBATCH -o output.txt # Mail me on job start & end #SBATCH --mail-user=your_email #SBATCH --mail-type=BEGIN,END cd $SLURM_SUBMIT_DIR module load amber/16 module load intel/2019 module load gcc/8.2.0 srun $AMBERHOME/bin/pmemd.MPI -O -i mdin.in -o mdout.out -p prmtop.prm -c inpcrd.rst -ref inpcrd.rst -x trjfile.trj -inf file.info -r file.rst7
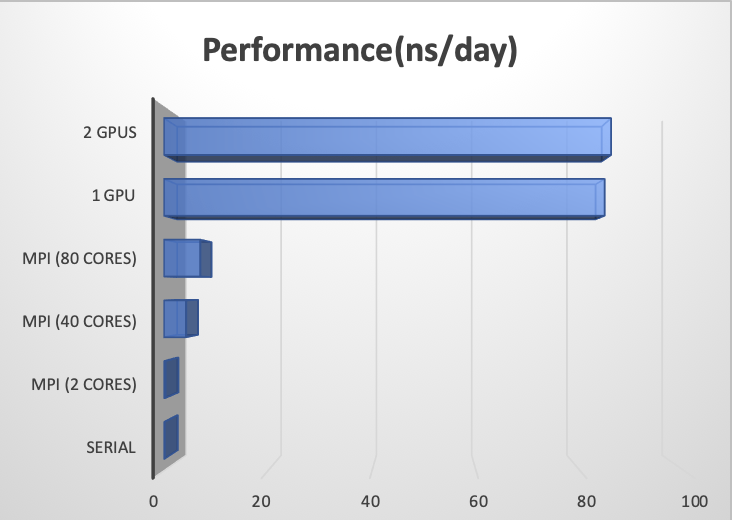
Benchmarks
Version: Amber 18
Dataset: Cellulose (408,609 atoms) NPT Ensemble
Merit: ns/day (the higher the better)
|
|
Performance(ns/day) |
|
Serial |
0.15 |
|
MPI (2 cores) |
0.28 |
|
MPI (40 cores) |
4.27 |
|
MPI (80 cores) |
6.99 |
|
1 GPU |
84.71 |
|
2 GPUs |
85.94 |
Additional Notes
To use Amber 18 load the relevant environment module: module load amber/18. Use pmemd.MPI for MPI-only version, pmemd.cuda for CUDA-only version, pmemd.cuda.MPI for CUDA+MPI version.
